This post is a tutorial explaining how to convert fMRI datasets from
4dfp to
NIfTI format. Most people not at Washington University in St Louis probably won't encounter a 4dfp dataset, since the format isn't in wide use. But much of the post isn't 4dfp-specific, and goes over ways to check (and correct) the orientation and
handedness of voxel arrays. The general steps and alignment tests here should apply, regardless of which image format is being converted to NIfTI.
Converting images between formats (and software programs) is relatively easy ... ensuring that the orientation is set properly in the converted images is sometimes not, and often irritating. But it is very, very worth your time to make sure that the initial image conversion is done properly; sorting out orientation at the end of an analysis is much harder than doing it right from the start.
To understand the issues, remember that NIfTI (and 4dfp, and most other image formats) have two types of information: the 3d or 4d array of voxel values, and how to interpret those values (such as how to align the voxel array to anatomical space, the size of the voxels in mm, etc.), which is stored in a header.
This post gives has more background information, as does
this one. The
NIfTI file specification allows several different ways of specifying alignment, and both
left and right-handed voxel arrays. Flexibility is good, I guess, but can lead to great confusion, because different programs have different strategies for interpreting the header fields. Thus, the exact same NIfTI image can look different (e.g. left/right flipped, shifted off center) when opened in different programs. That's what we want to avoid: we want to create NIfTI images that will be read with proper alignment into most programs.
Step 1: Read the image into R as a plain voxel array
I use the
R4dfp R package to read 4dfp images into
R, and the
oro.nifti package to (
read or) write NIfTI images out of R. The R4dfp package won't run in Windows, so needs to be run on a linux box, mac, or within
NeuroDebian (oro.nifti runs on all platforms).
in.4dfp <- R4dfp.Load(paste0(in.path, img.name));
vol <- in.4dfp[,,,];
The 4dfp image
fname at location
in.path is read into R with the first command, and the voxel matrix extracted with the second. You can check properties of the 4dfp image by checking its fields, such as
in.4dfp$dims and
in.4dfp$center. The dimension of the extracted voxel array should match the input 4dfp fields (
in.4dfp$dims == dim(vol)).
Step 2: Determine the handedness of the input image and its alignment
Now, look at the extracted voxel array: how is it orientated? My strategy is to open the original image in the program whose display I consider to be the ground truth, then ensure the matrix in R matches that orientation. For 4dfp images, the "ground truth" program is
fidl; often it's the program which created the input image. I then look at the original image and find a slice with a clear asymmetry, and display that slice in R with
image.
Here's slice k=23 (green arrow) of the first timepoint in the image (blue arrow). The greyscale image is the view in fidl which we consider true: the left anterior structure (white arrow) is "pointy". The R code
image(vol[,,23,1])) displays this same slice, using hot colors (bottom right). We can see that it's the correct slice (I truncated the side a bit in the screen shot), but the "pointy" section is at the bottom right of the image instead of the top left. So, the voxel array needs flipped left/right and anterior/posterior.
Step 3: Convert and flip the array as needed
This
doFlip R function reorders the voxel array. It takes and returns a 3d array, so should be run on each timepoint individually. This takes a minute or so to run, but I haven't bothered speeding it up, since it only needs to be run once per input image. If you have a faster version, please share!
# flip the data matrix in the specified axis direction(s). Be very careful!!!
doFlip <- function(inArray, antPost=FALSE, leftRight=FALSE) {
if (antPost == FALSE & leftRight == FALSE) { stop("no flips specified"); }
if (length(dim(inArray)) != 3) { stop(print("not three dimensions in the array")); }
outvol <- array(NA, dim(inArray));
IMAX <- dim(inArray)[1];
JMAX <- dim(inArray)[2];
KMAX <- dim(inArray)[3];
for (k in 1:KMAX) {
for (j in 1:JMAX) { # i <- 1; j <- 1; k <- 1;
for (i in 1:IMAX) {
if (antPost == TRUE & leftRight == FALSE) { outvol[i,j,k] <- inArray[i,(JMAX-j+1), k]; }
if (antPost == TRUE & leftRight == TRUE) { outvol[i,j,k] <- inArray[(IMAX-i+1),(JMAX-j+1), k]; }
if (antPost == FALSE & leftRight == TRUE) { outvol[i,j,k] <- inArray[(IMAX-i+1), j, k]; }
}
}
}
return(outvol);
}
# put the array to right-handed coordinate system, timepoint-by-timepoint.
outvox <- array(NA, dim(vol));
for (sl in 1:dim(vol)[4]) { outvox[,,,sl] <- doFlip(vol[,,,sl], TRUE, TRUE); }
image(outvox[,,23,1])

The last three code call the function (
TRUE, TRUE specifies flipping both left-right and anterior-posterior), putting the flipped timepoint images into the new array
outvox. Now, when we look at the same slice and timepoint with the image command (
image(outvox[,,23,1]))), it is orientated correctly.
Step 4: Write out as a NIfTI image, confirming header information
A 3 or 4d array can be written out as a NIfTI image without specifying the header information (e.g. with
writeNIfTI(nifti(outvox), fname), which uses the
oro.nifti defaults). Such a "minimal header" image can be read in and out of R just fine, but will almost certainly not be displayed properly when used as an overlay image in standard programs (e.g., on a template anatomy in
MRIcroN) - it will probably be out of alignment. The values in the NIfTI header fields are interpreted by programs like MRIcroN and the
Workbench to ensure correct registration.
Some of the
NIfTI header fields are still mysterious to me, so I usually work out the correct values for a particular dataset iteratively: writing a NIfTI with my best guess for the correct values, then checking the registration, then adjusting header fields if needed.
out.nii <- nifti(outvox, datatype=64, dim=dim(outvox), srow_x=c(3,0,0,in.4dfp$center[1]),
srow_y=c(0,3,0,in.4dfp$center[2]), srow_z=c(0,0,3,in.4dfp$center[3]))
out.nii@sform_code <- 1
out.nii@xyzt_units <- 2; # for voxels in mm
pixdim(out.nii)[1:8] <- c(1, 3,3,3, dim(outvox)[4], 1,1,1);
# 1st slot is qfactor, then mm, then size of 4th dimension.
writeNIfTI(out.nii, paste0(out.path, img.name))
These lines of code write out a 4d NIfTI with header values for my example image. The input image has 3x3x3 mm voxels, which is directly entered in the code, and the origin is read out of the input fields (you can also type the numbers in directly, such as
srow_x=c(3,0,0,72.3)). The image below shows how to confirm that the output NIfTI image is orientated properly: the value of the voxel under the crosshairs is the same (blue arrows) in the original 4dfp image in fidl, the NIfTI in R, and the NIfTI in MRIcroN. The red arrows point out that the voxel's i,j,k coordinates are the same in MRIcroN and R, and the left-right flipping matches (yellow arrow).
Here's another example of what a correctly-aligned file looks like: the NIfTI output image is displayed here as an overlay on a template anatomy in MRIcroN (overlay is red) and the Workbench (overlay is yellow). Note that the voxel value is correct (blue), and that the voxel's x,y,z coordinates (purple) match between the two programs. If you look closely, the voxel's i,j,k coordinates (blue) are 19,4,23 in MRIcroN (like they were in R), but 18,3,22 in the Workbench. This is ok:
Workbench starts counting from 0, R and MRIcroN from 1.
concluding remarks
It's straightforward to combine the R code snippets above into loops that convert an entire dataset. My strategy is usually to step through the first image carefully - like in this post - then convert the images for the rest of the runs and participants in a loop (i.e., everyone with the same scanning parameters). I'll then open several other images, confirming they are aligned properly.
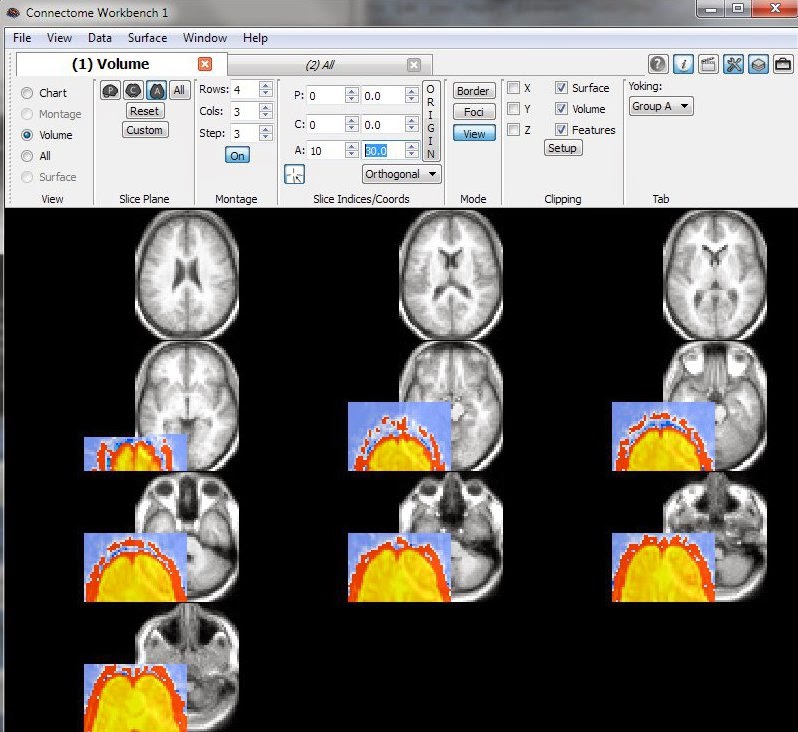
This last image is a concrete example of what you don't want to see: the functional data (colored overlay) is offset from the anatomical template (greyscale). Don't panic if you see this: dive back into the headers.
 At left is an example of a confusion matrix produced by a classifier, where the test set was balanced, with 10 examples of class face, and 10 of class place. The classifier did quite well: 9 of the 10 face examples were (correctly) labeled face, and 8 of the 10 place examples were (correctly) labeled place. For lack of a better term, what I'll call "regular" or "overall" accuracy is calculated as shown at left: the proportion of examples correctly classified, counting all four cells in the confusion matrix. Balanced accuracy is calculated as the average of the proportion corrects of each class individually. In this example, both the overall and balanced calculations produce the same accuracy (0.85), as will always happen when the test set has the same number of examples in each class.
At left is an example of a confusion matrix produced by a classifier, where the test set was balanced, with 10 examples of class face, and 10 of class place. The classifier did quite well: 9 of the 10 face examples were (correctly) labeled face, and 8 of the 10 place examples were (correctly) labeled place. For lack of a better term, what I'll call "regular" or "overall" accuracy is calculated as shown at left: the proportion of examples correctly classified, counting all four cells in the confusion matrix. Balanced accuracy is calculated as the average of the proportion corrects of each class individually. In this example, both the overall and balanced calculations produce the same accuracy (0.85), as will always happen when the test set has the same number of examples in each class. This second example is the same as the first, but I took out one of the place test examples: now the testing dataset was not balanced, as it has 10 face examples, but only 9 place examples. As you can see, now the overall accuracy is a bit higher than the balanced.
This second example is the same as the first, but I took out one of the place test examples: now the testing dataset was not balanced, as it has 10 face examples, but only 9 place examples. As you can see, now the overall accuracy is a bit higher than the balanced. 
























{kind=link}